Different Profiles of Microglial Activation in Alzheimer's disease
What's the science?
Microglia, the immune cells of the brain, may contribute to Alzheimer’s disease by becoming activated in response to brain pathology (also known as neuroinflammation). Currently, whether neuroinflammation is associated with Alzheimer’s progression (harms the brain) or whether it may be protective (helps to “eat” plaques in the brain) is still a matter of debate. This week in Brain, Hamelin and colleagues used PET imaging to examine how microglial activation in the brain is related to Alzheimer’s disease progression.
How did they do it?
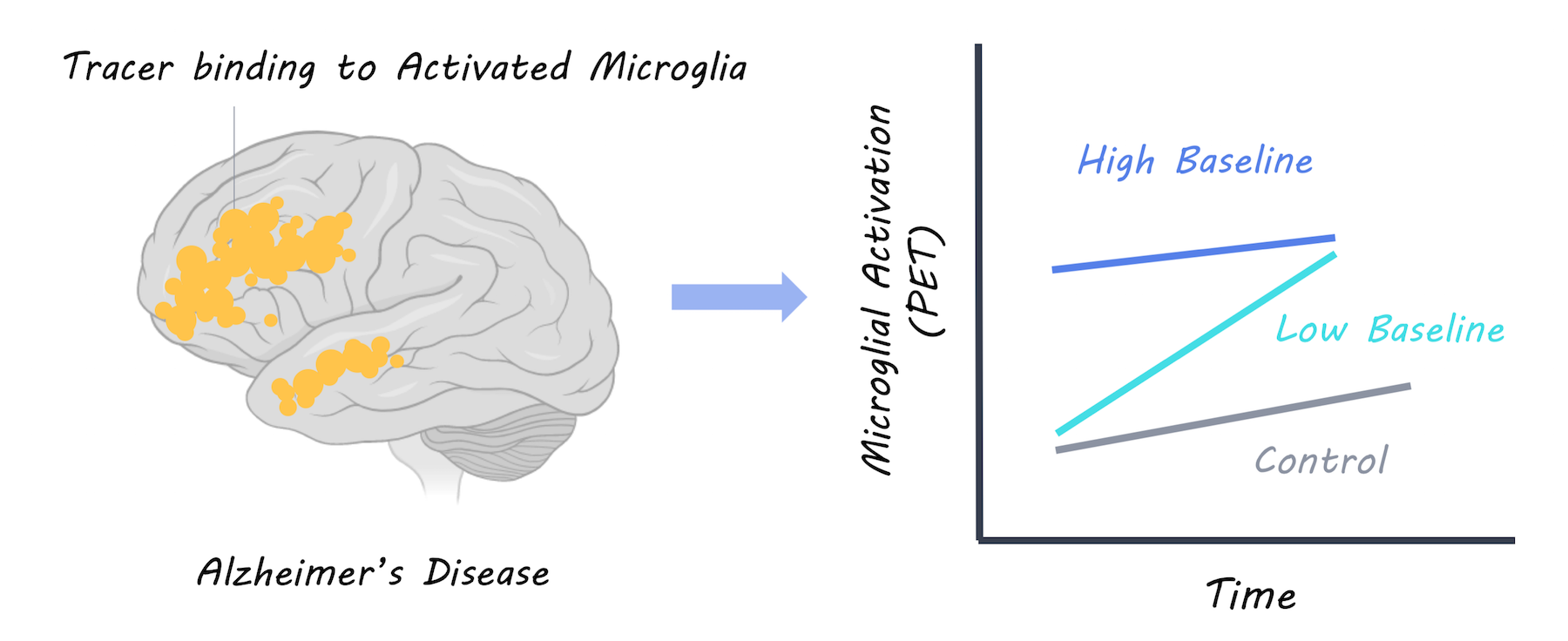
PET imaging was used to measure the uptake of a radiotracer (18F-DPA-714) in the brain binding to activated microglia. A large group of patients with Alzheimer’s disease were scanned twice for activated microglia, once at baseline and once two years later. They were then followed up annually and scanned with MRI to measure brain volume (measure of Alzheimer’s progression) and given annual cognitive tests to assess dementia severity and cognitive function. Based on this, patients were split into “fast and slow decliner” categories. The microglial activation levels over time were also analyzed compared to a control group of healthy participants.
What did they find?
Having a high level of microglial activation at baseline was predictive of being a slow decliner. In patients with a high baseline neuroinflammation, cognitive performance was better and brain volume was more preserved, suggesting that more microglial activation at baseline is protective. At two years follow-up, microglial activation was higher in Alzheimer’s participants but not controls as would be expected. Increased microglial activation over time was related to worsening cognitive scores and brain atrophy, suggesting that it is harmful. However, when they examined neuroinflammation over time at an individual level, they found that those with the highest baseline microglial activation had the lowest increase in microglial activation over time. They concluded that there is a dynamic relationship, whereby neuroinflammation may affect patients differently, depending on their original level of microglial activity. Microglial activation appears to be protective initially, but exacerbates Alzheimer’s disease over time; to a greater extent in those who had low levels of microglial activation to begin with.
What's the impact?
This is the first study to show that neuroinflammation may affect individuals with Alzheimer’s disease differently depending on their baseline level of microglial activity. It shows us that microglial activation may be helpful or harmful depending on the individual and how far their disease has progressed. Understanding the role microglial activation play in Alzheimer’s disease is an essential part of understanding how the disease progresses.
L. Hamelin et al., Distinct dynamic profiles of microglial activation are associated with progression of Alzheimer's disease. Brain (2018). Access the original scientific publication here.