On-Demand Gene Therapy for Brain Circuit Disorders
Post by Andrew Vo
The takeaway
Current gene therapies that aim to correct abnormal neuronal activity in different disorders fail to discern between pathological and surrounding healthy neurons. Designing therapies that are activity-dependent allows treatments to be delivered specifically to misfiring neurons in a more targeted manner.
What's the science?
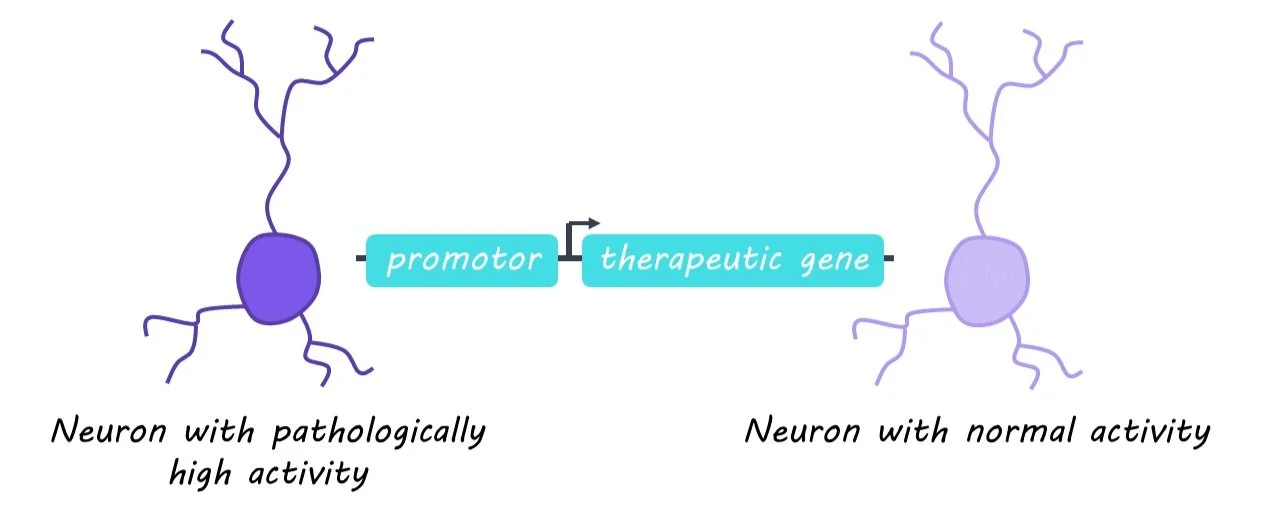
Genetic therapies can augment and treat pathological neuronal activity associated with many neurodevelopmental and neuropsychiatric disorders. However, current options fail to discern between pathological and surrounding healthy neurons, increasing the risk of unwanted side effects. Activity-dependent promoters (gene sequences that only activate following neuronal activity and control the expression of delivered therapeutic genes) could be the key to targeting pathologically overactive neurons. This week in Science, Qiu, et al. test whether an activity-dependent promoter can reduce seizures and seizure-related activity in a model of epilepsy.
How did they do it?
The authors began with cell cultures in which they introduced either (1) a therapeutic gene that encodes potassium channels and decreases neuronal excitability or (2) a control gene that did not affect neuronal activity. The expression of both genes fell under the control of an activity-dependent promoter. They then measured whether their therapeutic gene reduced hyperexcitability. Next, the authors delivered either the therapeutic or control gene into the hippocampus of adult mice. A drug was used to induce generalized seizures and then hippocampal neuronal excitability between the two groups was recorded and compared. In another experiment, mice were injected with a second consecutive dose of the seizure-inducing drug to test whether initial activation of the therapeutic gene had lasting effects. The authors evaluated the mice on different behavioral tests of learning and working memory. Finally, they tested the effectiveness of their activity-dependent therapy in a mouse model of chronic epilepsy with spontaneous seizures, and in neurons derived from human stem cells.
What did they find?
The authors found that cell cultures treated with the therapeutic gene showed reduced neuronal excitability. Closer inspection of the cells revealed that the promoter was selectively activated in excitatory but not inhibitory neurons, which indicates that the therapeutic gene was specific to neurons involved in pathological overexcitability. Similarly, the excitability of activated neurons in response to drug-induced seizures was decreased in the hippocampus of mice treated with the therapeutic gene. When a second dose was injected after a short delay, the authors noted greatly reduced seizures. This suggests that activation of the therapeutic gene following the initial seizure persisted and protected against the second seizure. This benefit was time-limited, however, as a third dose injected after a longer delay when therapeutic gene expression had returned to baseline was no longer attenuated. No significant changes in behavior were observed between the therapeutic and control gene groups. These collective findings were also replicated in a mouse model of chronic epilepsy and human neurons.
What's the impact?
The present study describes a gene therapy for epilepsy that decreases neuronal excitability in an activity-dependent manner. This approach was time-limited to the duration of pathological neuronal activity, effective in mouse models of both discrete and chronic epilepsy, and translatable to human neurons. Activity-dependent gene therapy is an exciting and promising strategy in the targeted treatment of brain disorders.