
Synaptic Transcription Is Driven by Circadian Rhythm While Protein Expression Is Driven by Sleep in the Mouse Forebrain
Post by Amanda McFarlan
What's the science?
The sleep-wake cycle is involved in regulating the size of synapses and brain protein expression, so, it is not surprising that it may also be involved in regulating local synaptic transcription and protein expression. In support of this, it has been shown previously that approximately 6% of transcription in the forebrain fluctuates throughout the day, which is thought to be mediated by the sleep-wake cycle. This week in Science, Noya and colleagues investigated the role of sleep and circadian rhythms on the rhythmicity of synaptic transcript and protein expression in the mouse forebrain and found a remarkably different picture than that seen in transcripts in the cell as a whole.
How did they do it?
The authors collected tissue from the mouse forebrain every 4 hours for one 24-hour period to assess daily rhythms in messenger RNA (mRNA) transcripts and protein expression. They used biochemical homogenization and fractionation to purify synaptoneurosomes (isolated synaptic terminals) and identify mRNAs that were present in these synapses. Then, using isolated synaptoneurosomes, they performed mass spectrometry-based proteomics to identify patterns of protein expression in the synaptic proteome (i.e. all proteins expressed in the synapse) and the total forebrain. Next, the authors investigated the role of the sleep-wake cycle in regulating transcription of synaptic mRNA and protein expression by collecting forebrain tissue every 4 hours for one 24-hour period from mice that were sleep-deprived for four hours prior to sacrifice. They assessed the effect of high sleep pressure (i.e. sleep deprivation/greater amplitude of delta oscillations) on the daily rhythmicity of mRNA and protein expression in forebrain tissue.
What did they find?
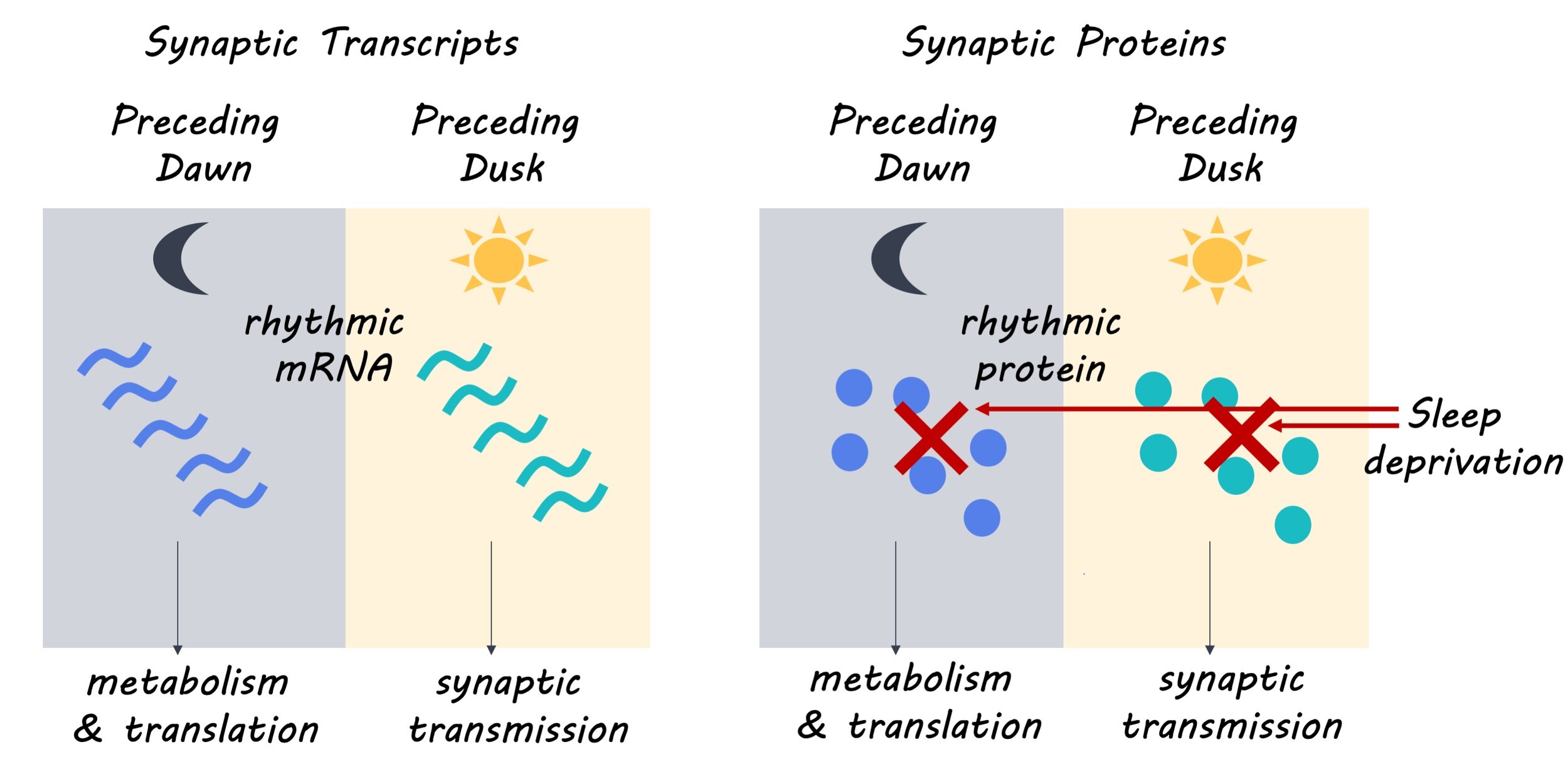
The authors found that 67% of synaptic mRNA transcripts had a rhythmic pattern of expression, with 93% of these transcripts exclusively showing rhythmic patterns of expression in synaptoneurosomes. This suggests that the oscillatory or cyclic pattern of synaptic mRNA expression may be controlled by post-transcriptional mechanisms. They also found that 11.7% of synaptic proteins and 17.2% of proteins in the forebrain showed rhythmic patterns of expression. Next, the authors determined that the rhythmic expression of synaptic mRNA formed two distinct clusters separating mRNA transcripts expressed in the day from those expressed at night. Furthermore, they found that the mRNA transcripts expressed in the day participated in biological processes including synapse organization and transmission, while mRNA transcripts expressed at night participated in biological processes such as metabolism, cell proliferation and development. These distinct patterns of expression and biological roles were also observed in the synaptic proteome, with 75% of rhythmically expressed synaptic proteins showing concomitance with their mRNA counterpart. Finally, the authors determined that the rhythmicity of synaptic mRNA transcript expression was mostly preserved with high sleep pressure, but rhythmicity in protein expression could no longer be detected with high sleep pressure, suggesting that synaptic mRNA expression may be controlled by a molecular clock while protein expression may be gated by sleep-wake pressure.
What's the impact?
This is the first study to show that synaptic mRNA transcripts in the mouse forebrain have a highly rhythmic expression that is controlled post-transcriptionally. The rhythmic expression of mRNA transcripts and their related proteins was shown to be important for distinct biological processes associated with either day or night. These findings provide insight into the mechanisms by which synaptic mRNA and protein expression are regulated in the mouse forebrain.

Noya et al. The forebrain synaptic transcriptome is organized by clocks but its proteome is driven by sleep (2019). Access the original scientific publication here.




